Flujo de trabajo para análisis de genomas¶
Autoras: M. Nayeli Luis-Vargas y Marisol Navarro
Objetivo
Conocer el flujo de trabajo general que se lleva a cabo en un análisis genomico, así como las principales herramientas bionformáticas utilizadas en cada paso.
Comprender los archivos input y output conseguidos en cada paso del proceso.
Cuando hablamos de un análisis genómico, nos referimos al ensamble del genoma de un organismo. Después de secuenciar nuestras muestras, recibimos un archivo de texto plano con los fragmentos de DNA (que llamamos lecturas) correspondientes al organismo en cuestión. Estos fragmentos deben ser ensamblados, es decir, unidos como si se tratará de armar un rompecabezas, con el fin de conocer la composición genética del organismo que nos interesa. A partir de un genoma ensamblado podemos saber que tipo de genes tiene, para qué protéina codifican o podemos comparar su composición genética con la de otro organismo (organimos de referencia) y así identificar variaciones.
Los fragmentos de DNA obtenidos a partir del secuenciador son conocidos como lecturas y cada plataforma de secuenciación produce distitnos tamaños de estos.
El proceso para ensamblar un genoma se muestra en la siguiente figura (da click en ésta para que puedas visualizarla mejor):
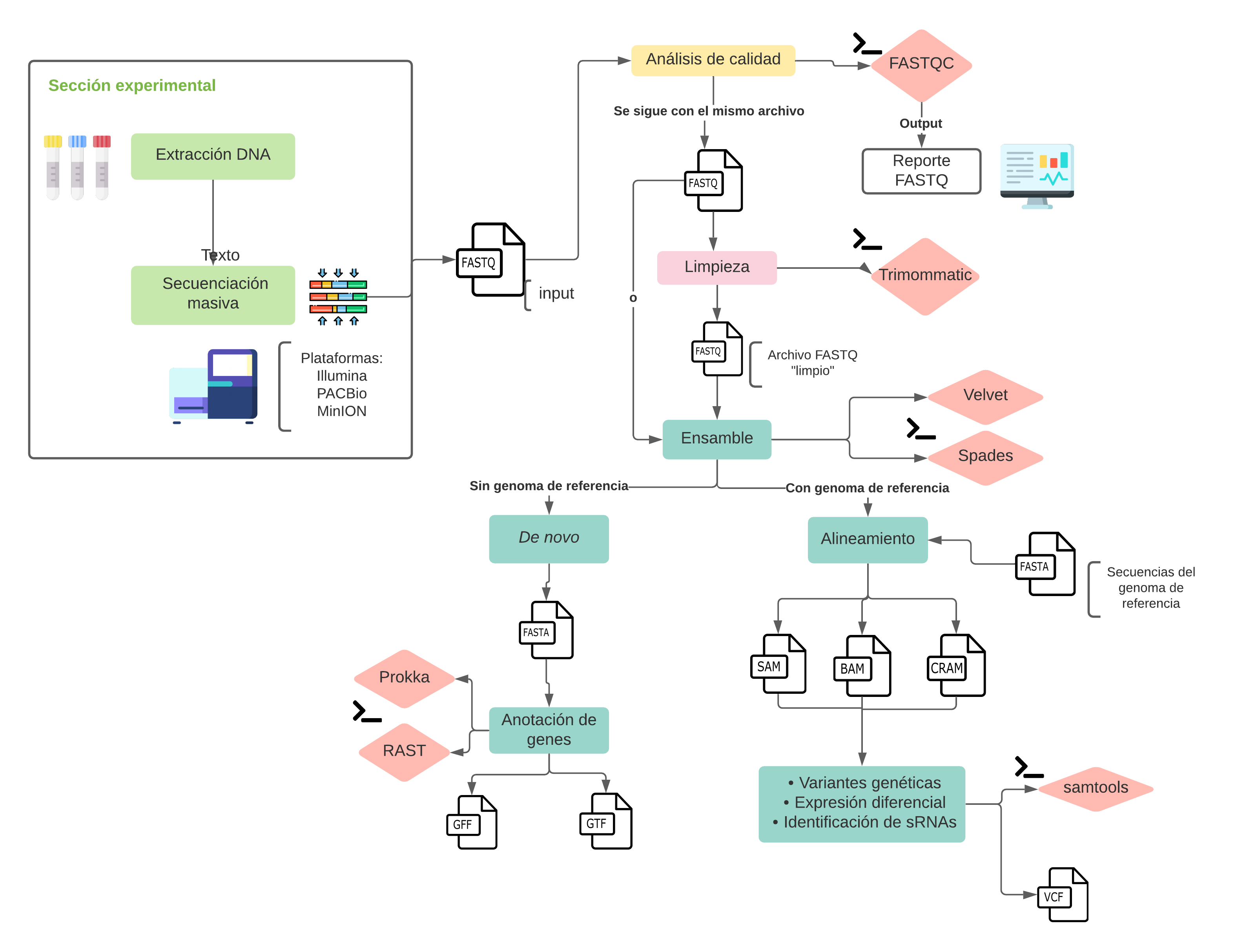
Fig. 28 Flujo de trabajo en un análisis genómico.¶
Análisis de calidad¶
Archivos
Archivo input: Archivo .fastq obtenido del secuenciador.
Archivo output: No hay archivo como tal, sino un reporte en HTML que indica la calidad de lase secuencas.
El proceso de secuenciación puede tener algunos errores, algunos de éstos pueden ser:
La variación en la preparación de la muestra
Algun tipo de contaminación
Duplicados por PCR
Recombinación
Amplificación selectiva
Error por sustitución de bases
Inserciones y deleciones
De manera que es importante saber si nuestras secuencias tienen una calidad aceptable para poder continuar con su análisis. El software màs utilizado para llevar a cabo éste proceso es FASTQC.
FastQC¶
Tiene como objetivo proporcionar una manera simple de hacer algunas comprobaciones de control de calidad de datos de secuencias procedentes de metodologías de secuenciación de alto rendimiento. En el software se ingresa un archivo .fastq y a partir de éste arroja una reporte en HTML el cual indica si los datos tienen algún problema de calidad que deba ser tomado en cuenta antes de realizar cualquier análisis posterior.
Las principales funciones de FastQC son:
Proporcionar una visión general y rápida que identifique en qué áreas puede haber problemas.
Gráficos y tablas para evaluar rápidamente los datos.
Operación fuera de línea para permitir la generación automática de informes sin ejecutar la aplicación de forma interactiva.
Intepretación del reporte de FASTQC
Para saber cómo se interpretan los resultados del reporte arrojado por FASTQC puedes ver el vídeo Using FastQC to check the quality of high throughput sequence
Limpieza¶
Archivos
Archivo input: Archivo .fastq obtenido del secuenciador.
Archivo output: Archivo .fastq “limpio”.
Durante éste paso se eliminan los adaptadores que se colocaron durante la secuenciación, así mismo pueden eliminarse secuencias sobrerrepresentadas o hacer algún corte en donde la secuencia haya reflejado una mala calidad. Existen diversas herrameintas para llevar a cabo esta limpieza desde un archivo .fastq. Algunas de éstas herramientas son:
Ensamble¶
Después de los procesos mencionados (calidad y limpieza de las secuencas) el ensamble del genoma puede llevarse a cabo de dos maneras: a partir de un genoma de referencia o sin un genoma de referencia, es decir de novo. ¿En qué se diferencian? Imagina que estás armando un rompecabezas, si tienes la imagen del rompecabezas (la que viene en la caja), todo será más sencillo, puedes puedes ir comparando las piezas con la imagen y darte una idea. Ahora, imagina que no tienes la imagen…Se vuelve más complicado porque no tienes idea de cómo luce, ni que figuras tiene, ni los patrones ni nada de nada. Si estuvieras en éste caso, pues tendrás que buscar otras estrategias.
Ensamble sin genoma de referencia: de novo¶
Archivos
Archivo input: Archivo .fastq “limpio”.
Archivo output: Archivo .fasta.
En general, el ensamble de novo se basa en la simple suposición de que los fragmenos de DNA (las lecturas) con bastante similitud se originan de la misma posición dentro del genoma. De ésta manera, la similitud entre secuencias de DNA se usa para conectar fragmentos individuales en secuencias contiguas más largas, denominadas contigs (secuencias consenso obtenidas a partir del ensamblaje de los fragmentos de DNA).
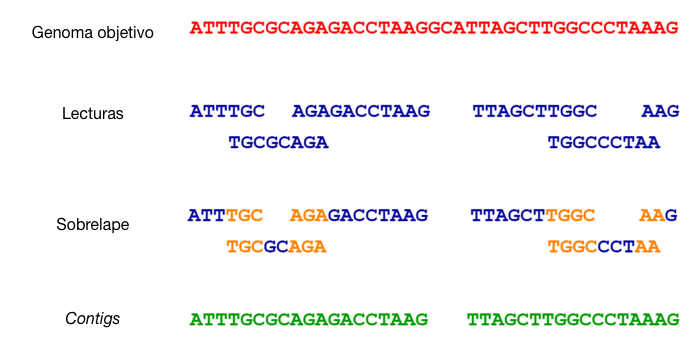
Fig. 29 Proceso general en un ensamble de novo.¶
En el ensamble de novo las lecturas de corta longitud (como las que se obtienen a partir de tecnologías de secuenciación masiva) constituyen un problema cuando hay repeticiones en el genoma, es decir, segmentos de DNA que aparecen más de una vez a lo largo del genoma. Cuando una lectura proviene de una región repetitiva, y es más corto que la región completa, no se sabe con certeza de cuál copia de la repetición se obtuvo. Es por ello que durante el ensamblaje, se pueden crear falsas uniones en las regiones de repetición. Para evitar este problema se han empleado algunas estrategias computacionles, éstas son:
OLC (*Overlap-Layout-Consesus*)Éste método identifica todos los pares de lecturas que se solapan lo suficientemente bien y organiza ésta información en un grafo (una red), en la cual hay uno nodo por cada par de lecturas de ellos y un enlace. Ésta estructura permite que se tome en cuenta la relación global entre las lecturas. De ésta manera, se definen caminos que corresponde con los segmentos del genoma que están siendo ensamblados. Se reconstruye el genoma mediante la búsqueda de un único camino que atraviese todos los nodos de una sola vez [1].
Gráficos De BrujinDescompone las lecturas en k-meros, los k-meros son subcadenas de longitud k contenidas dentro de una secuencia biológica. De manera que se modela la relación entre subcadenas exactas de logitud k dentro de las lecturas. En el grafo de De Brujin los nodos son k-meros y los enlaces indican qué k-meros adyacentes se solapan por k-1 letras, por lo que la longitud del k-mero se correlaciona con la longitud del solapamiento que el ensamblador es capaz de detectar. La mayoría de los ensambladores usan la información global de las lecturas para refinar la estructura del gráfico, resolver repeticiones y eliminar patrones no consistentes. Además, incorporan métodos de corrección de errores para mejorar la calidad del ensamblaje [1].
Software para ensamble de genomas de novo¶
Los softwares más utilizados para el ensamble son Velvet y SPAdes. Sin embargo puedes consultar ésta página para consultar todas las herramientas que actualmente existen para ensamblar un genoma.
Ensamble con genoma de referencia¶
Archivos
Archivos input: Archivo .fastq “limpio” y archivo .fasta que es el genoma de referencia.
Archivo output: Archivo .sam/.bam/.cram.
Cuando se tiene un genoma de referencia, se hace un mapeo o alineamiento de los fragmentos de DNA de nuestra muestras con un genoma de referencia, es decir, con un genoma previamente ensamblado y cuyas secuencias podemos descargar del NCBI o que podemos obtener de estudios anteriores. A partir de éste mapeo se hace el ensamble. Cuando se hace un ensamble de éste tipo, uno de los objetivos puede ser comparar ambos genomas, ¿En qué genes se diferencian? y hacer un estudio conocido como llamado de variantes genéticas variant calling. La llamada de variantes implica identificar polimorfismo de un solo nucleótido (SNPs) y pequeñas inserciones y deleciones de los datos.
Anotación de genes¶
Archivos
Archivo input: Archivo .fasta.
Archivo output: Archivo .gff/.gtf.
La anotación de genes es el proceso de identificar cualquier elemento funcional a lo largo de la secuencia. La anotación da significado al genoma al proporcionar la ubicación y la función de genes y regiones reguladoras. Los programas más utilizados actualmente para anotar genes en procariontes son Prokka y RAST.
Recursos recomendados